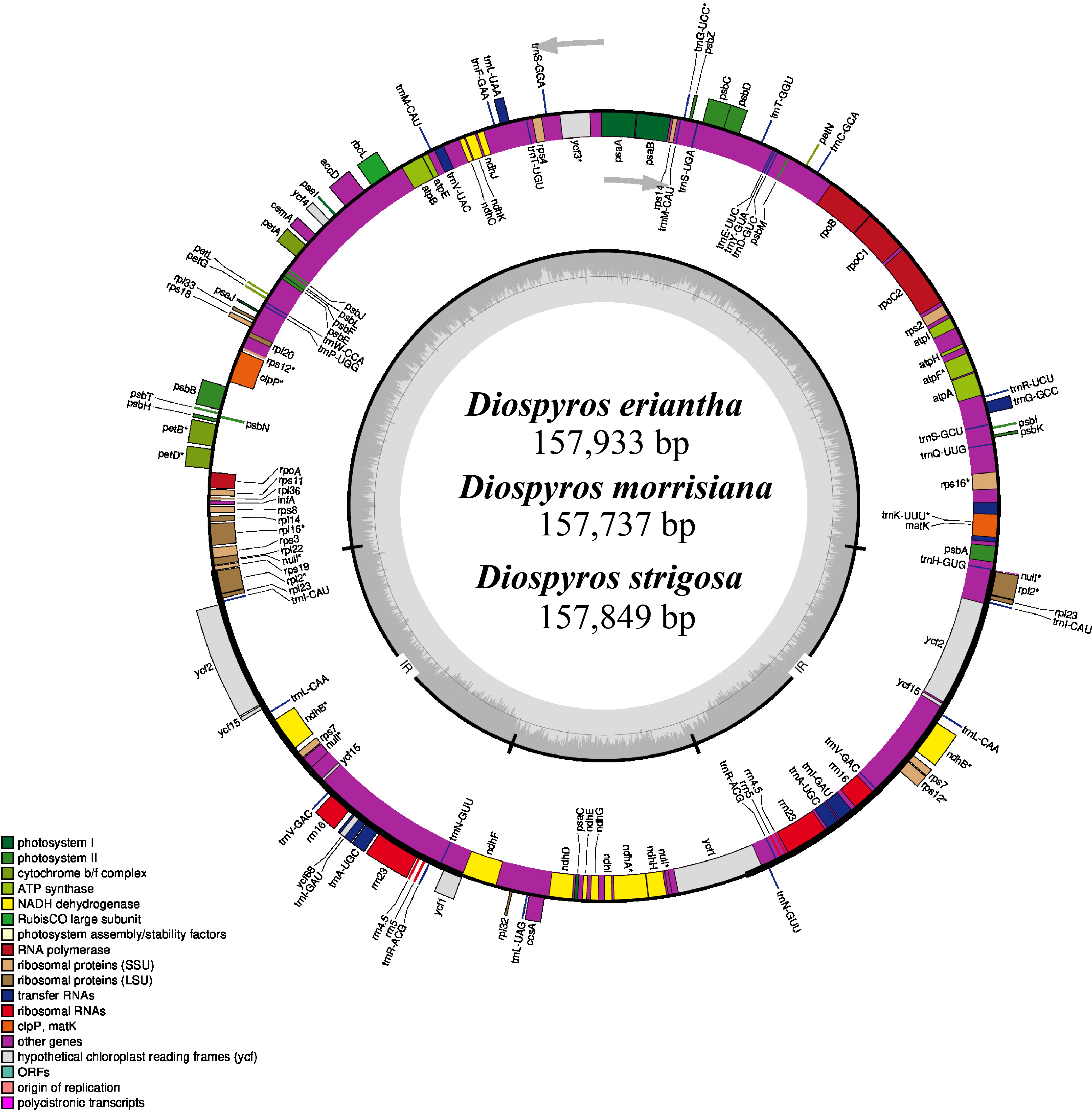
Diospyros (Ebenaceae) is a widely distributed genus of trees and shrubs native to tropical and subtropical regions, with numerous species valued for their fruits (persimmons), timber, and medicinal values. However, information regarding their plastomes and chloroplast evolution is scarce. The present study performed comparative genomic and evolutionary analyses on plastomes of 18 accepted Diospyros species, including three newly sequenced ones. Our study showed a highly conserved genomic structure across the species, with plastome size ranging from 157,321 bp (D. jinzaoshi) to 157,934 bp (D. deyangensis). These plastomes encoded 134–138 genes, including 89–91 protein-coding genes, 1–2 pseudogenes (Ψycf1 for all, Ψrps19 for a few), 37 tRNA genes, and 8 rRNA genes. Comparative analysis of Diospyros identified the intergenic regions (trnH-psbA, rps16-trnQ, trnT-psbD, petA-psbJ, trnL-trnF-ndhJ) as the mutational hotspots in these species. Phylogenomic analyses identified three main groups within the genus designated as the evergreen, deciduous, and island groups. The codon usage analysis identified 30 codons with relative synonymous codon usage (RSCU) values greater than 1 and 29 codons ending with A and U bases. A total of three codons (UUA, GCU, and AGA) with highest (RSCU) values were identified as the optimal codons. ENC-plot indicated the significant role of mutational pressure in shaping codon usage, while most protein-coding genes in Diospyros experienced relaxed purifying selection (Ka/Ks < 1). Additionally, the ndhG, rpoC1, and ycf3 genes showed positive selection (Ka/Ks > 1) in the island, deciduous, and both deciduous and evergreen species, respectively. Thus, the results provide a foundation for elaborating Diospyros’s genetic architecture and taxonomy, conserving genetic diversity and enriching genetic resources.