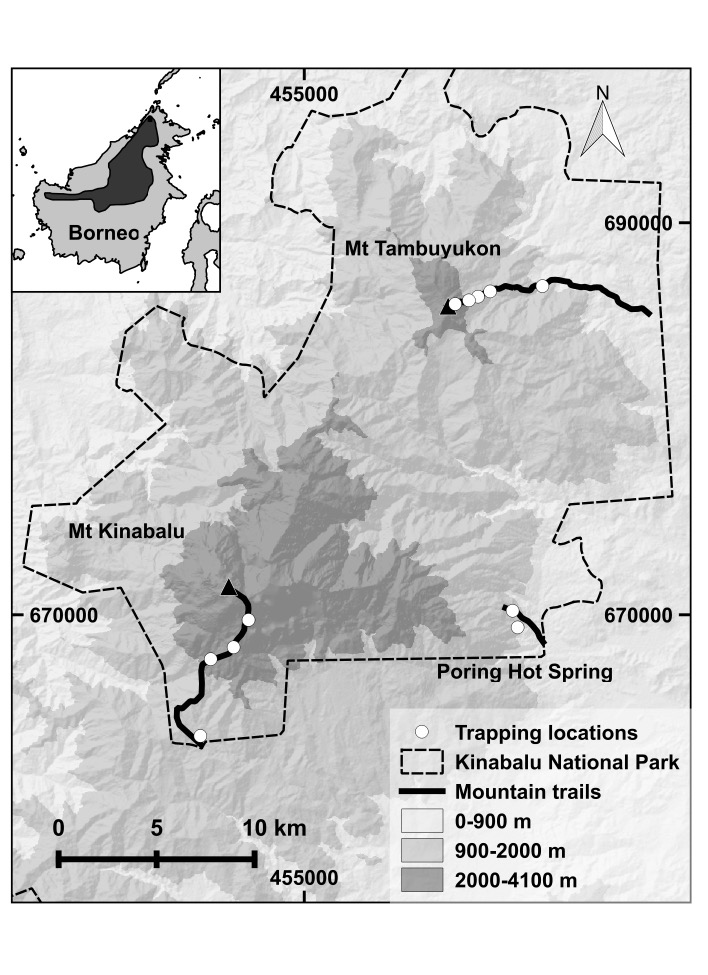
Rapid shifts in environmental variables associated with elevational changes in montane ecosystems provide opportunities to test hypotheses regarding the effects of environmental heterogeneity on gene flow and genetic structure. In tropical mountains, spatial environmental heterogeneity combined with seasonal environmental stability is predicted to result in low dispersal across elevations. Few studies have investigated the genetic consequences of elevational environmental heterogeneity in tropical montane mammals. Here, we use a population genomics approach to test the hypothesis that mountain treeshrews (Tupaia montana) exhibit limited gene flow across elevational gradients and between two neighboring peaks within Kinabalu National Park (KNP) in Borneo. We sampled 83 individuals across elevations on Mt. Tambuyukon (MT) and Mt. Kinabalu (MK) and sequenced mitogenomes and 4,106 ultraconserved elements containing an average of 1.9 single nucleotide polymorphisms per locus. We detected high gene flow across elevations and between peaks. We found greater genetic differentiation on MT than MK despite its lower elevation and associated environmental variation. This implies that, contrary to our hypothesis, genetic structure in this system is not primarily shaped by elevation. We propose that this pattern may instead be the result of colonization history combined with restricted upslope gene flow on MT due to unique plant communities associated with its upper montane habitats. Our results serve as a foundation to identify and mitigate future effects of climate change on mountain treeshrews in KNP. Given predictions for 2100 CE, we predict that mountain treeshrews will maintain genetic connectivity in KNP, making it an important conservation stronghold.