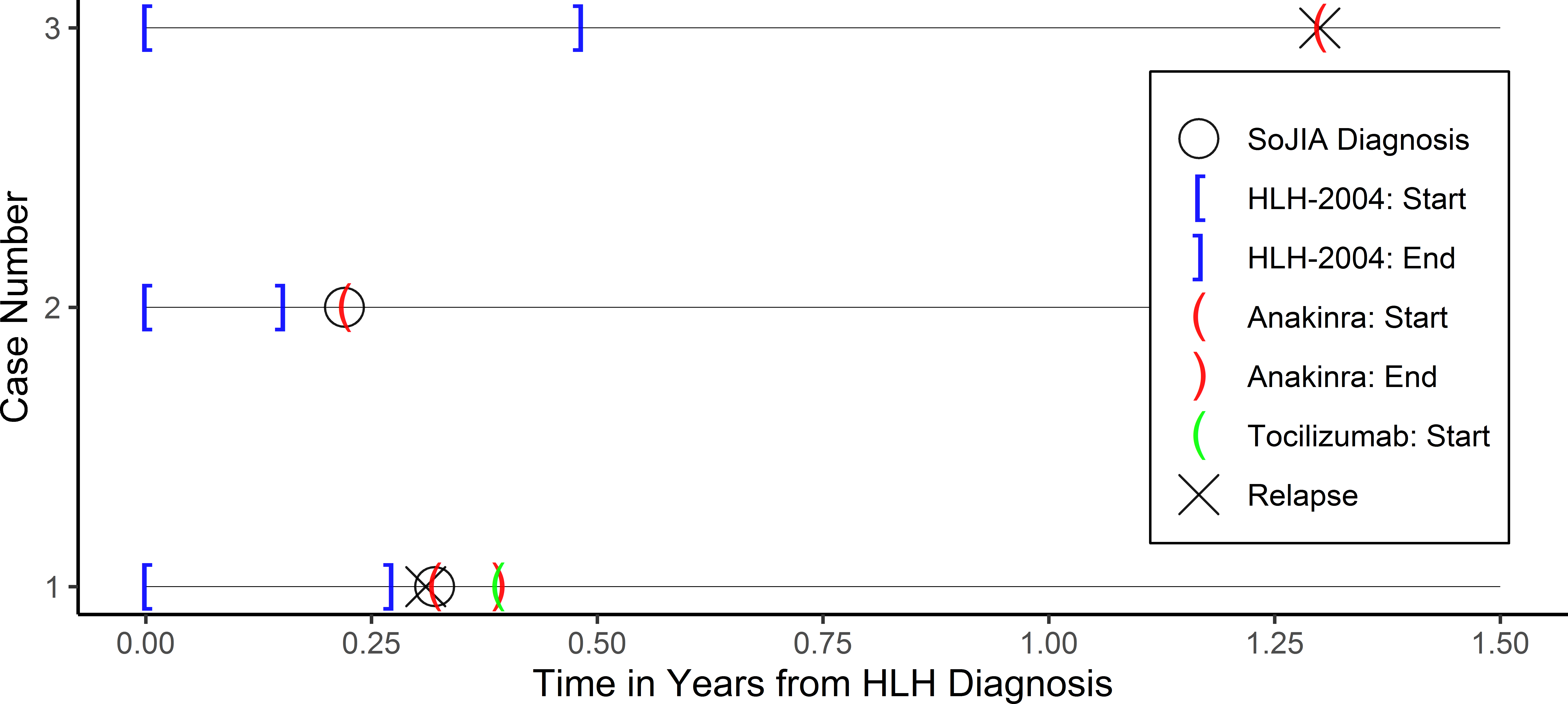
Comment on: The use of anakinra in the treatment of secondary hemophagocytic lymphohistiocytosis1Paige Vicenzi, OMS-IV, 2Zahra Jiwani, DO, 3Ricardo Guirola, MD,1,4Tyler Hamby, PhD, 5Anish Ray, MD1Texas College of Osteopathic Medicine, University of North Texas Health Science Center2Department of Pediatrics, Children’s Hospital of Michigan3Department of Pediatric Rheumatology, Cook Children’s Health Care System4Department of Research Operations, Cook Children’s Health Care System5Department of Pediatric Hematology/Oncology, Cook Children’s Health Care SystemCorresponding Author:Anish Ray, MD1500 Cooper St., 5th floor,Fort Worth, TX 76104Phone: [email protected] Count: 497Number of Tables: 0Number of Figures: 1Running Title: Anakinra in Secondary HLHKeywords: hemophagocytic lymphohistiocytosis, anakinra, pediatricThe authors have no financial support or conflicts of interest.Hemophagocytic lymphohistiocytosis (HLH) is a rare yet potentially fatal systemic disease arising from uncontrolled activation of the immune system. According to the Histiocyte Society’s 2004 guidelines, patients must meet five of eight criteria to be diagnosed with HLH [1]. HLH may be classified into primary and secondary. Primary, or familial, HLH is attributed to underlying defects in genes that control natural killer (NK) and cytotoxic T-lymphocyte (CTL) cell degranulation. Secondary HLH, in contrast, may occur in the context of triggers, such as malignancy, rheumatologic disease, or infection. Systemic-onset juvenile idiopathic arthritis (SoJIA) is a well-recognized trigger of HLH and both share overlapping features (e.g. fever and elevated ferritin). Management of SoJIA includes the immunomodulator Anakinra, an interleukin 1 (IL-1) receptor antagonist hypothesized to dampen an overactive immune system. Three patients treated for HLH with concomitant SoJIA diagnosis at Cook Children’s Medical Center between 2014 and 2019 are described below in order to examine the role of immunomodulators in their clinical course and outcome.Three Hispanic patients (aged 8-15) presented with a constellation of systemic symptoms, including fever, generalized rash, fatigue, and weight loss. Upon fulfilment of necessary criteria and subsequent diagnosis of HLH, they were treated accordingly with HLH-2004 protocol. Case 3, whose HLH was suspected to be secondary to Epstein-Barr Virus (EBV) infection, rapidly responded to treatment and, therefore, briefly discontinued Etoposide; however, she tolerated this poorly and resumed treatment after a six-week hiatus with the addition of weekly Rituximab to mitigate rising EBV titers. All patients achieved remission.Past medical history for case 3 included autoimmune disorders such as celiac disease, type 1 diabetes and suspected idiopathic juvenile arthritis for which she did not require ongoing care prior to presenting with features of secondary HLH. Cases 1 and 2 were diagnosed with SoJIA following their HLH diagnosis. Cases 1 and 3 relapsed with HLH within months of their initial encounter. Due to their concurrent diagnosis of SoJIA, both received daily Anakinra. Case 3 experienced rapid resolution of symptoms. In contrast, Case 1 had unsatisfactory response of musculoskeletal manifestations prompting switch from Anakinra to weekly Tocilizumab—another biologic that antagonizes IL-6 receptor—with favorable response. Case 2 was started on daily Anakinra immediately following his diagnosis of SoJIA and has yet to relapse. In summary, all cases have yet to experience an additional relapse following introduction of Anakinra or Tocilizumab. Figure 1 provides the treatment timelines for Cases 1-3 who had 5.37, 2.87, and 4.62 years of follow up, respectively.Though traditional therapy for HLH includes intensive courses of etoposide and corticosteroids with substantial risk for morbidity and mortality, biologics represent a newer class of medications highly effective in treating diseases with inflammatory or immune-mediated components [2]. In a reimaging of the HLH treatment algorithm, a recent study proposes Anakinra as initial treatment with sequential escalation of immunosuppression to mitigate adverse effects [3]. This case series reinforces that immunomodulators, such as Anakinra, are safe and promising treatment options in pediatric patients with secondary HLH.